510(k) summarya summary, submitted under section 513(i) of the act, of the safety and effectiveness information contained in a premarket notification submission upon which a determination of substantial equivalence can be based. Safety and effectiveness information refers to safety and effectiveness data and information supporting a finding of substantial equivalence, including all adverse safety and effectiveness information.
[FDA 21CFR807]
abnormal useconscious, intentional act or intentional omission of an act that is counter to or violates normal use and is also beyond any further reasonable means of user interface-related risk control by the manufacturer
Examples: Reckless use or sabotage or intentional disregard of information for safety are such acts.
Note 1: An intended but erroneous action that is not abnormal use is considered a type of use error.
Note 2: Abnormal use does not relieve the manufacturer from considering non-user interface-related means of risk control.
Note 3: The figure below shows the relationships of the types of use.
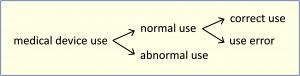
[IEC 62366-1:2015]
accessorya finished device that is intended to support, supplement, and/or augment the performance of one or more parent devices
[FDA guidance: Medical Device Accessories...(2017)]
accessoryadditional part for use with equipment in order to:
– achieve the intended use,
– adapt it to some special use,
– facilitate its use,
– enhance its performance, or
– enable its functions to be integrated with those of other equipment
[IEC 60601-1:2012]
accessory for a medical devicean article which, whilst not being itself a medical device, is intended by its manufacturer to be used together with one or several particular medical device(s) to specifically enable the medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the medical device(s) in terms of its/their intended purpose(s)
[EU MDR 2017/745]
accessory for an in vitro diagnostic medical devicean article which, whilst not being itself an
in vitro diagnostic medical device, is intended by its manufacturer to be used together with one or several particular in vitro diagnostic medical device(s) to specifically enable the
in vitro diagnostic medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the
in vitro diagnostic medical device(s) in terms of its/their intended purpose(s)
[EU IVDR 2017/746]
accompanying documentationmaterials accompanying a medical device and containing information for the user or those accountable for the installation, use, maintenance, decommissioning and disposal of the medical device, particularly regarding safe use
Note 1: The accompanying documentation can consist of the instructions for use, technical description, installation manual, quick reference guide, etc.
Note 2: Accompanying documentation is not necessarily a written or printed document but could involve auditory, visual, or tactile materials and multiple media types.
[ISO 14971:2019]
active deviceany device, the operation of which depends on a source of energy other than that generated by the human body for that purpose, or by gravity, and which acts by changing the density of or converting that energy. Devices intended to transmit energy, substances or other elements between an active device and the patient, without any significant change, shall not be deemed to be active devices
Note: Software shall also be deemed to be an active device
[EU MDR 2017/745]
adulterated drugs and devices[paraphrased] A drug or device shall be deemed to be adulterated...if
(a) Poisonous, insanitary, etc., ingredients; adequate controls in manufacture...
(b) Strength, quality, or purity differing from official compendium...
(c) Misrepresentation of strength, etc., where drug is unrecognized in compendium...
(d) Mixture with or substitution of another substance...
(e) Devices not in conformity with performance standards...
(f) Certain class III devices...
(g) Banned devices...
(h) Manufacture, packing, storage, or installation of device not in conformity with applicable requirements or conditions...
(i) Failure to comply with requirements under which device was exempted for investigational use...
(j) Delayed, denied, or limited inspection; refusal to permit entry or inspection...
[OLRC 21USC9 section 351 excerpt]
Note: Actual definition is too long to list here.
amortizationan accounting technique used to periodically lower the book value of a loan or intangible asset over a set period of time
Note: The term "amortization" can refer to two situations. First, amortization is used in the process of paying off debt through regular principal and interest payments over time. An amortization schedule is used to reduce the current balance on a loan, for example a mortgage or car loan, through installment payments. Second, amortization can also refer to the spreading out of capital expenses related to intangible assets over a specific duration – usually over the asset's useful life – for accounting and tax purposes.
[Investopedia]
analytechemical substance that is the subject of chemical analysis
[ISO 11139:2018]
analytical performanceability of an IVD medical device to detect or measure a particular analyte
[ISO 20916:2019]
analytical performancethe ability of a device to correctly detect or measure a particular analyte
[EU IVDR 2017/746]
ARCI chart (RACI chart)chart describing the participation by various roles when completing tasks and/or deliverables
auditsystematic, independent and documented process for obtaining objective evidence and evaluating it objectively to determine the extent to which the audit criteria are fulfilled
Note 1: The fundamental elements of an audit include the determination of the conformity of an object according to a procedure carried out by personnel not being responsible for the object audited.
Note 2: An audit can be an internal audit (first party), or an external audit (second party or third party), and it can be a combined audit or a joint audit.
Note 3: Internal audits, sometimes called first-party audits, are conducted by, or on behalf of, the organization itself for management review and other internal purposes, and can form the basis for an organization’s declaration of conformity. Independence can be demonstrated by the freedom from responsibility for the activity being audited.
Note 4: External audits include those generally called second and third-party audits. Second party audits are conducted by parties having an interest in the organization, such as customers, or by other persons on their behalf. Third-party audits are conducted by external, independent auditing organizations such as those providing certification/registration of conformity or governmental agencies.
[ISO 9000:2015]
benefitpositive impact or desirable outcome of the use of a medical device on the health of an individual, or a positive impact on patient management or public health
Note: Benefits can include positive impact on clinical outcome, the patient’s quality of life, outcomes related to diagnosis, positive impact from diagnostic devices on clinical outcomes, or positive impact on public health.
[ISO 14971:2019]
benefit-risk determinationthe analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose, when used in accordance with the intended purpose given by the manufacturer
[EU MDR 2017/745, EU IVDR 2017/746]
bill of materials (BOM)an extensive list of raw materials, components, and instructions required to construct, manufacture, or repair a product or service
Note: A bill of materials usually appears in a hierarchical format, with the highest level displaying the finished product and the bottom level showing individual components and materials
[Investopedia]
biological producta virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, protein, or analogous product, or arsphenamine or derivative of arsphenamine (or any other trivalent organic arsenic compound), applicable to the prevention, treatment, or cure of a disease or condition of human beings.
(1) A virus is interpreted to be a product containing the minute living cause of an infectious disease and includes but is not limited to filterable viruses, bacteria, rickettsia, fungi, and protozoa.
(2) A therapeutic serum is a product obtained from blood by removing the clot or clot components and the blood cells.
(3) A toxin is a product containing a soluble substance poisonous to laboratory animals or to man in doses of 1 milliliter or less (or equivalent in weight) of the product, and having the property, following the injection of non-fatal doses into an animal, of causing to be produced therein another soluble substance which specifically neutralizes the poisonous substance and which is demonstrable in the serum of the animal thus immunized.
(4) An antitoxin is a product containing the soluble substance in serum or other body fluid of an immunized animal which specifically neutralizes the toxin against which the animal is immune.
(5) A product is analogous:
(i) To a virus if prepared from or with a virus or agent actually or potentially infectious, without regard to the degree of virulence or toxicogenicity of the specific strain used.
(ii) To a therapeutic serum, if composed of whole blood or plasma or containing some organic constituent or product other than a hormone or an amino acid, derived from whole blood, plasma, or serum.
(iii) To a toxin or antitoxin, if intended, irrespective of its source of origin, to be applicable to the prevention, treatment, or cure of disease or injuries of man through a specific immune process.
(6) A protein is any alpha amino acid polymer with a specific, defined sequence that is greater than 40 amino acids in size. When two or more amino acid chains in an amino acid polymer are associated with each other in a manner that occurs in nature, the size of the amino acid polymer for purposes of this paragraph (h)(6) will be based on the total number of amino acids in those chains, and will not be limited to the number of amino acids in a contiguous sequence.
[FDA 21CFR600]
business risk managementmedical device risk management that focuses on loss of revenue or profits to the business
CE markinga marking by which a manufacturer indicates that a device is in conformity with the applicable requirements set out in this Regulation and other applicable Union harmonisation legislation providing for its affixing
[EU MDR 2017/745, EU IVDR 2017/746]
change controlassessment and determination of the appropriateness of a proposed alteration to product, process, or equipment
[ISO 11139:2018]
classone of the three categories of regulatory control for medical devices, defined below:
(1) Class I means the class of devices that are subject to only the general controls authorized by or under sections 501 (adulteration), 502 (misbranding), 510 (registration), 516 (banned devices), 518 (notification and other remedies), 519 (records and reports), and 520 (general provisions) of the act. A device is in class I if (i) general controls are sufficient to provide reasonable assurance of the safety and effectiveness of the device, or (ii) there is insufficient information from which to determine that general controls are sufficient to provide reasonable assurance of the safety and effectiveness of the device or to establish special controls to provide such assurance, but the device is not life-supporting or life-sustaining or for a use which is of substanial importance in preventing impairment of human health, and which does not present a potential unreasonable risk of illness of injury.
(2) Class II means the class of devices that is or eventually will be subject to special controls. A device is in class II if general controls alone are insufficient to provide reasonable assurance of its safety and effectiveness and there is sufficient information to establish special controls, including the promulgation of performance standards, postmarket surveillance, patient registries, development and dissemination of guidance documents (including guidance on the submission of clinical data in premarket notification submissions in accordance with section 510(k) of the act), recommendations, and other appropriate actions as the Commissioner deems necessary to provide such assurance. For a device that is purported or represented to be for use in supporting or sustaining human life, the Commissioner shall examine and identify the special controls, if any, that are necessary to provide adequate assurance of safety and effectiveness and describe how such controls provide such assurance.
(3) Class III means the class of devices for which premarket approval is or will be required in accordance with section 515 of the act. A device is in class III if insufficient information exists to determine that general controls are sufficient to provide reasonable assurance of its safety and effectiveness or that application of special controls described in paragraph (c)(2) of this section would provide such assurance and if, in addition, the device is life-supporting or life-sustaining, or for a use which is of substantial importance in preventing impairment of human health, or if the device presents a potential unreasonable risk of illness or injury.
[FDA 21CFR860]
classification namethe term used by the Food and Drug Administration and its classification panels to describe a device or class of devices for purposes of classifying devices under section 513 of the act
[FDA 21CFR807]
cleaningphysical removal of soil and contaminants from an item to the extent necessary for further processing or for the intended use
[FDA guidance: Reprocessing Medical Devices...(2017)]
cleaningremoval of contaminants to the extent necessary for further processing or for intended use
[ISO 11139:2018]
clinical benefitthe positive impact of a device on the health of an individual, expressed in terms of a meaningful, measurable, patient-relevant clinical outcome(s), including outcome(s) related to diagnosis, or a positive impact on patient management or public health
[EU MDR 2017/745]
clinical benefitthe positive impact of a device related to its function, such as that of screening, monitoring, diagnosis or aid to diagnosis of patients, or a positive impact on patient management or public health
[EU IVDR 2017/746]
clinical datainformation concerning safety or performance that is generated from the use of a device and is sourced from the following:
— clinical investigation(s) of the device concerned,
— clinical investigation(s) or other studies reported in scientific literature, of a device for which equivalence to the device in question can be demonstrated,
— reports published in peer reviewed scientific literature on other clinical experience of either the device in question or a device for which equivalence to the device in question can be demonstrated,
— clinically relevant information coming from post-market surveillance, in particular the post-market clinical follow-up
[EU MDR 2017/745]
clinical evaluationassessment and analysis of clinical data pertaining to a medical device to verify the clinical safety and performance of the device when used as intended by the manufacturer
[ISO 13485:2016]
clinical evaluationsystematic and planned process to continuously generate, collect, analyse and assess the clinical data pertaining to a device in order to verify the safety and performance, including clinical benefits, of the device when used as intended by the manufacturer
[EU MDR 2017/745]
clinical evidenceclinical data and clinical evaluation results pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer
[EU MDR 2017/745]
clinical evidenceclinical data and performance evaluation results, pertaining to a device of a sufficient amount and quality to allow a qualified assessment of whether the device is safe and achieves the intended clinical benefit(s), when used as intended by the manufacturer
[EU IVDR 2017/746]
clinical investigationany systematic investigation involving one or more human subjects, undertaken to assess the safety or performance of a device
[EU MDR 2017/745]
clinical investigation (clinical research, clinical study)any experiment that involves a test article and one or more human subjects, and that either must meet the requirements for prior submission to the Food and Drug Administration under section 505(i) or 520(g) of the act, or need not meet the requirements for prior submission to the Food and Drug Administration under these sections of the act, but the results of which are intended to be later submitted to, or held for inspection by, the Food and Drug Administration as part of an application for a research or marketing permit. The term does not include experiments that must meet the provisions of part 58, regarding nonclinical laboratory studies.
[FDA 21CFR50, FDA 21CFR56]
Note: The terms research, clinical research, clinical study, study, and clinical investigation are deemed to be synonymous for purposes of this part.
[FDA 21CFR56]
clinical investigation (clinical study, clinical trial)systematic investigation in one or more human subjects, undertaken to assess the clinical performance, effectiveness or safety of a medical device
Note: For the purpose of this document, “clinical trial” or “clinical study” are synonymous with “clinical investigation”.
[ISO 14155:2020]
clinical performancethe ability of a device, resulting from any direct or indirect medical effects which stem from its technical or functional characteristics, including diagnostic characteristics, to achieve its intended purpose as claimed by the manufacturer, thereby leading to a clinical benefit for patients, when used as intended by the manufacturer
[EU MDR 2017/745]
clinical performancethe ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user
[EU IVDR 2017/746]
clinical performancebehaviour of a medical device and response of the subject(s) to that medical device in relation to its intended use, when correctly applied to appropriate subject(s)
Note: Clinical performance can be defined under national regulations.
[ISO 14155:2020]
clinical performance of an IVD medical deviceability of an IVD medical device to yield results that are correlated with a particular clinical condition or physiological/pathological process/state in accordance with the intended use (clinical test purpose, target population and intended user)
Note: In accordance with intended use, clinical performance can include expected values, diagnostic sensitivity and diagnostic specificity based on the known clinical condition or physiological/pathological process/state of the individual, and negative and positive predictive values based on the prevalence of the disease.
[ISO 20916:2019]
clinical performance studystudy undertaken to establish or confirm the clinical performance of an IVD medical device
[ISO 20916:2019]
combination productincludes:
(1) A product comprised of two or more regulated components, i.e., drug/device, biologic/device, drug/biologic, or drug/device/biologic, that are physically, chemically, or otherwise combined or mixed and produced as a single entity;
(2) Two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products;
(3) A drug, device, or biological product packaged separately that according to its investigational plan or proposed labeling is intended for use only with an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product the labeling of the approved product would need to be changed, e.g., to reflect a change in intended use, dosage form, strength, route of administration, or significant change in dose; or
(4) Any investigational drug, device, or biological product packaged separately that according to its proposed labeling is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
[FDA 21CFR3]
Note 1: "Co-packaged combination product" has the meaning set forth in §3.2(e)(2) of this chapter.
Note 2: "Single-entity combination product" has the meaning set forth in §3.2(e)(1) of this chapter.
[FDA 21CFR4]
combination productentity presented as a single health care product that physically, chemically, or otherwise brings together or mixes items regulated under separate legislation
[ISO 11139:2018]
commercial distributionany distribution of a device intended for human use which is held or offered for sale but does not include the following:
(1) Internal or interplant transfer of a device between establishments within the same parent, subsidiary, and/or affiliate company;
(2) Any distribution of a device intended for human use which has in effect an approved exemption for investigational use under section 520(g) of the act and part 812 of this chapter;
(3) Any distribution of a device, before the effective date of part 812 of this chapter, that was not introduced or delivered for introduction into interstate commerce for commercial distribution before May 28, 1976, and that is classified into class III under section 513(f) of the act: Provided, That the device is intended solely for investigational use, and under section 501(f)(2)(A) of the act the device is not required to have an approved premarket approval application as provided in section 515 of the act; or
(4) For foreign establishments, the distribution of any device that is neither imported nor offered for import into the United States.
[FDA 21CFR807]
complaintwritten, electronic or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, usability, safety or performance of a medical device that has been released from the organization's control or related to a service that affects the performance of such medical devices
[ISO 13485:2016]
complaintany written, electronic, or oral communication that alleges deficiencies related to the identity, quality, durability, reliability, safety, effectiveness, or performance of a device after it is released for distribution
[FDA 21CFR820]
componentany raw material, substance, piece, part, software, firmware, labeling, or assembly which is intended to be included as part of the finished, packaged, and labeled device.
[FDA 21CFR820]
conformity (conformance)fulfilment of a requirement
[ISO 9000:2015]
conformity assessmentthe process demonstrating whether the requirements of this Regulation relating to a device have been fulfilled
[EU MDR 2017/745, EU IVDR 2017/746]
conformity assessment bodya body that performs third-party conformity assessment activities including calibration, testing, certification and inspection
[EU MDR 2017/745, EU IVDR 2017/746]
contract manufacturer[supplier that] manufactures a finished device to another establishment's specifications
Note: Includes contract packagers
[FDA website: Who Must Register...]
contract sterilizer[supplier that] provides a sterilization service for another establishment's devices
[FDA website: Who Must Register...]
copyrighta form of protection provided to the authors of "original works of authorship" including literary, dramatic, musical, artistic, and certain other intellectual works, both published and unpublished.
Note: The 1976 Copyright Act generally gives the owner of copyright the exclusive right to reproduce the copyrighted work, to prepare derivative works, to distribute copies or phonorecords of the copyrighted work, to perform the copyrighted work publicly, or to display the copyrighted work publicly.
[USPTO]
correct usenormal use without use error
Note 1: Deviation from instructions for use is only considered use error if it leads to a medical device response that is different than intended by the manufacturer or expected by the user.
Note 2: The figure below shows the relationships of the types of use.
[IEC 62366-1:2015]
correctionaction to eliminate a detected nonconformity
[ISO 9000:2015, ISO 11139:2018]
corrective actionaction to eliminate the cause of a nonconformity and to prevent recurrence
[ISO 9000:2015]
corrective actionaction taken to eliminate the cause of a potential or actual non-conformity or other undesirable situation
[EU MDR 2017/745, EU IVDR 2017/746]
corrective and preventive action (CAPA)The purpose of the corrective and preventive action subsystem is to collect information, analyze information, identify and investigate product and quality problems, and take appropriate and effective corrective and/or preventive action to prevent their recurrence. Verifying or validating corrective and preventive actions, communicating corrective and preventive action activities to responsible people, providing relevant information for management review, and documenting these activities are essential in dealing effectively with product and quality problems, preventing their recurrence, and preventing or minimizing device failures.
[FDA website: Guide to Inspections of Quality Systems]
custom devicea device within the meaning of section 520(b) of the Federal Food, Drug, and Cosmetic Act
[FDA 21CFR812]
custom-made deviceany device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person's professional qualifications which gives, under that person's responsibility, specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their individual conditions and needs
Note: However, mass-produced devices which need to be adapted to meet the specific requirements of any professional user and devices which are mass-produced by means of industrial manufacturing processes in accordance with the written prescriptions of any authorised person shall not be considered to be custom-made devices
[EU MDR 2017/745]
customerperson or organization that could or does receive a product or a service that is intended for or required by this person or organization
Example: Consumer, client, end-user, retailer, receiver of product or service from an internal process, beneficiary and purchaser.
Note: A customer can be internal or external to the organization.
[ISO 9000:2015]
customer satisfactioncustomer’s perception of the degree to which the customer’s expectations have been fulfilled
Note 1: It can be that the customer’s expectation is not known to the organization, or even to the customer in question, until the product or service is delivered. It can be necessary for achieving high customer satisfaction to fulfil an expectation of a customer even if it is neither stated nor generally implied or obligatory.
Note 2: Complaints are a common indicator of low customer satisfaction but their absence does not necessarily imply high customer satisfaction.
Note 3: Even when customer requirements have been agreed with the customer and fulfilled, this does not necessarily ensure high customer satisfaction.
[ISO 9000:2015]
customer serviceinteraction of the organization with the customer throughout the life cycle of a product or a service
[ISO 9000:2015]
de novo classification requesta submission requesting
de novo classification under section 513(f)(2) of the Federal Food, Drug, and Cosmetic Act
[FDA 21CFR4]
depreciationan accounting method of allocating the cost of a tangible or physical asset over its useful life or life expectancy
Note: Depreciation represents how much of an asset's value has been used up. Depreciating assets helps companies earn revenue from an asset while expensing a portion of its cost each year the asset is in use.
[Investopedia]
design and development plan (DDP)overall plan for design and development activities for the particular medical device under consideration
design changechange (modification) to product design or operations process design
design controlsprocess that increases the likelihood that the product design transferred to production translates into a medical device that meets its intended use (and user needs)
design freezedeclaration that verification and validation activities are very likely to succeed using the current Device Master Record
design history file (DHF)a compilation of records which describes the design history of a finished device
[FDA 21CFR820]
design inputthe physical and performance requirements of a device that are used as a basis for device design
[FDA 21CFR820]
design outputthe results of a design effort at each design phase and at the end of the total design effort. The finished design output is the basis for the device master record. The total finished design output consists of the device, its packaging and labeling, and the device master record.
[FDA 21CFR820]
Note: Design outputs can be divided into two categories:
1. Product Design Outputs (PDO) - design outputs pertaining to product design (i.e., medical device specifications, labeling specifications, packaging specifications)
2. Operations Design Outputs (ODO) - design outputs pertaining to operations process design (i.e., production process specifications, delivery process specifications, servicing process specifications)
design review (DR)a documented, comprehensive, systematic examination of a design to evaluate the adequacy of the design requirements, to evaluate the capability of the design to meet these requirements, and to identify problems
[FDA 21CFR820]
design transferthe sum of activities that ensure that the device design is correctly translated into production specification
design validationestablishing by objective evidence that device specifications conform with user needs and intended use(s)
[FDA 21CFR820]
design verificationconfirmation that the design output meets the design input requirements
[FDA 21CFR820 derived]
device history record (DHR)a compilation of records containing the production history of a finished device
[FDA 21CFR820]
device master record (DMR)a compilation of records containing the procedures and specifications for a finished device
[FDA 21CFR820]
DIOVVDesign Inputs, Design Outputs, Verification, and Validation
disinfectiona process that destroys pathogens and other microorganisms by physical or chemical means. Disinfection processes do not ensure the same margin of safety associated with sterilization processes.
The lethality of the disinfection process may vary, depending on the nature of the disinfectant (See Appendix D), which leads to the following subcategories:
a. High Level Disinfection: A lethal process utilizing a sterilant under less than sterilizing conditions. The process kills all forms of microbial life except for large numbers of bacterial spores.
b. Intermediate Level Disinfection: A lethal process utilizing an agent that kills viruses, mycobacteria, fungi and vegetative bacteria, but no bacterial spores.
c. Low Level Disinfection: A lethal process utilizing an agent that kills vegetative forms of 33 bacteria, some fungi, and lipid viruses.
[FDA guidance: Reprocessing Medical Devices...(2017)]
disinfectionprocess to inactivate viable microorganisms to a level previously specified as being appropriate for a defined purpose
[ISO 11139:2018]
distribution controlsmeasures that ensure that the packaged medical device and associated components are handled, stored and transported such that product design specifications are preserved with identification and traceability maintained throughout distribution
documentinformation and the medium on which it is contained
Example: Record, specification, procedure document, drawing, report, standard.
Note 1: The medium can be paper, magnetic, electronic or optical computer disc, photograph or master sample, or combination thereof.
Note 2: A set of documents, for example specifications and records, is frequently called “documentation”.
Note 3: Some requirements (e.g. the requirement to be readable) relate to all types of documents. However there can be different requirements for specifications (e.g. the requirement to be revision controlled) and for records (e.g. the requirement to be retrievable).
[ISO 9000:2015]
drug(A) articles recognized in the official United States Pharmacopoeia, official Homoeopathic Pharmacopoeia of the United States, or official National Formulary, or any supplement to any of them; and (B) articles intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease in man or other animals; and (C) articles (other than food) intended to affect the structure or any function of the body of man or other animals; and (D) articles intended for use as a component of any article specified in clause (A), (B), or (C). A food or dietary supplement for which a claim, subject to sections 343(r)(1)(B) and 343(r)(3) of this title or sections 343(r)(1)(B) and 343(r)(5)(D) of this title, is made in accordance with the requirements of section 343(r) of this title is not a drug solely because the label or the labeling contains such a claim. A food, dietary ingredient, or dietary supplement for which a truthful and not misleading statement is made in accordance with section 343(r)(6) of this title is not a drug under clause (C) solely because the label or the labeling contains such a statement.
[OLRC 21USC9]
effectiveThere is reasonable assurance that a device is effective when it can be determined, based upon valid scientific evidence, that in a significant portion of the target population, the use of the device for its intended uses and conditions of use, when accompanied by adequate directions for use and warnings against unsafe use, will provide clinically significant results.
[FDA 21CFR860]
effectivenessextent to which planned activities are realized and planned results are achieved
[ISO 9000:2015]
emergency usethe use of a test article on a human subject in a life-threatening situation in which no standard acceptable treatment is available, and in which there is not sufficient time to obtain IRB approval
[FDA 21CFR56]
essential output (EO)product design output that is essential for the proper functioning of the medical device
expected life of a devicethe time that a device is expected to remain functional after it is placed into use
Note: Certain implanted devices have specified “end of life” (EOL) dates. Other devices are not labeled as to their respective EOL, but are expected to remain operational through activities such as maintenance, repairs, or upgrades, for an estimated period of time.
[FDA 21CFR803]
expected service lifetime period specified by the manufacturer during which the medical device is expected to remain safe for use (i.e. maintain basic safety and essential performance)
Note: Maintenance can be necessary during the expected service life.
[IEC 62366-1:2015]
expensesthe cost of operations that a company incurs to generate revenue.
Note: Common expenses include payments to suppliers, employee wages, factory leases, and equipment depreciation.
[Investopedia]
experta person competent on the basis of appropriate education, training, skills and experience
[ISO/TR 24971:2020]
extended functional output (EFO)material, energy, or information that is delivered during use from a product that is outside the medical system but within the extended medical system in direct response to one or more functional outputs from the medical system
failure criticalitycombination of the severity of an effect and the frequency of its occurrence or other attributes of a failure as a measure of the need for addressing and mitigation
[IEC 60812:2006]
failure modemanner in which an item fails
[IEC 60812:2006]
failure mode and effects analysis (FMEA)a technique by which the consequences of an individual failure mode are systematically identified and evaluated
Note 1: FMEA is an inductive technique using the question “What happens if ... ?”. Components are analysed one at a time, thus generally looking at a single-fault condition. This is done in a “bottom-up” mode, i.e. following the procedure to the next higher functional system level.
[ISO/TR 24971:2020]
Note 2: FMEA is commonly divided into three types:
1. Design FMEA (DFMEA) – FMEA that considers failure modes associated with product design (i.e., assuming product meets product design specifications and product is used as intended)
2. Application FMEA (AFMEA) – FMEA that considers failure modes associated with product use (i.e., assuming product meets product design specifications but product is subject to use errors)
3. Process FMEA (PFMEA) – FMEA that considers failure modes associated with the production process or other operations process
failure mode effects and criticality analysis (FMECA)extension to FMEA that includes a means of ranking the severity of failure modes to allow prioritization of countermeasures
Note: Analogous to FMEA, the three types of FMECA are: DFMECA, AFMECA, and PFMECA.
fault tree analysis (FTA)an organized graphical representation of the conditions or other factors causing or contributing to the occurrence of a defined undesired outcome, referred to as the "top event"
Note 1: Fault tree analysis is a deductive (top-down) method of analysis aimed at pinpointing the causes or combinations of causes that can lead to the defined top event. The analysis can be qualitative or quantitative, depending on the scope of the analyses.
Note 2: When the outcome is a success, then the fault tree becomes a success tree, where the input events are those that contribute to the top success event.
[IEC 61025:2006 modified]
finished deviceany device or accessory to any device that is suitable for use or capable of functioning, whether or not it is packaged, labeled, or sterilized
[FDA 21CFR820]
Note: A device that is a constituent part of a combination product is considered a finished device within the meaning of the QS regulation.
[FDA 21CFR4]
foreign exporter[organization that] exports or offers for export to the United States (U.S.), a device manufactured, prepared, propagated, compounded, or processed in a foreign country, including devices originally manufactured in the United States. A foreign exporter must have an establishment address outside the U.S.
[FDA website: Who Must Register...]
fully refurbishingfor the purposes of the definition of manufacturer, means the complete rebuilding of a device already placed on the market or put into service, or the making of a new device from used devices, to bring it into conformity with this Regulation, combined with the assignment of a new lifetime to the refurbished device
[EU MDR 2017/745, EU IVDR 2017/746]
functional analysis (FA)systematic identification of the functional structure of a medical system, including system boundaries (interfaces), functional inputs, and functional outputs
functional inputmaterial, energy, or information that is delivered to a medical device or medical system during use
Note: Functional inputs can be divided into two categories:
1. Intended Functional Inputs - functional inputs inherent to the intended use
2. Unintended Functional Inputs - functional inputs not inherent to the intended use (i.e., environmental disturbances delivered to the medical device or medical system)
functional outputmaterial, energy, or information that is delivered from a medical device or medical system during use
Note: Functional outputs can be divided into two categories:
1. Intended Functional Outputs - functional outputs inherent to the intended use
2. Unintended Functional Outputs - functional outputs not inherent to the intended use (i.e., environmental disturbances delivered from the medical device or medical system)
harminjury or damage to the health of people, or damage to property or the environment
[ISO 14971:2019]
hazardpotential source of harm
[ISO 14971:2019]
Note: Basic hazards are directly associated with functional outputs. Extended hazards are directly associated with extended functional outputs (i.e., indirectly associated with functional outputs).
Hazard Analysis and Critical Control Point (HACCP)a systematic approach to identify hazards and hazardous situations and to control and monitor the associated risks by focusing on the critical control points in a manufacturing process
[ISO/TR 24971:2020]
hazard cause table (HCT)table for documenting the causes of hazards and exposure to hazards and the corresponding risk control measures
hazardous situationcircumstance in which people, property or the environment is/are exposed to one or more hazards
[ISO 14971:2019]
health care practitioner (HCP)a physician or other individual who is a health care provider and licensed under state law to prescribe drugs or devices
[FDA 21CFR99]
health care productmedical device, including
in vitro diagnostic medical device, or medicinal product, including biopharmaceutical
[ISO 11139:2018]
health productmedical device; drug; biologic product; human cell, tissue, and cellular and tissue-based product (HCT/P); or a combination product
health professionals practitioners, including physicians, nurses, pharmacists, dentists, respiratory therapists, physical therapists, technologists, or any other practitioners or allied health professionals that have a role in using a device for human use
[FDA 21CFR810]
health professionals practitioners, including physicians, nurses, pharmacists, dentists, respiratory therapists, physical therapists, technologists, or any other practitioners or allied health professionals that have a role in using a device for human use
[FDA 21CFR810]
health risk managementmedical device risk management that focuses on harm and safety as defined by ISO 14971
health risk managementmedical device risk management that focuses on harm and safety as defined by ISO 14971
human cell, tissue, and cellular and tissue-based products (HCT/P)HCT/Ps refers to human cell, tissue, and cellular and tissue-based products, as defined in §1271.3(d) of this chapter. An HCT/P that is not solely regulated under section 361 of the Public Health Service Act may be a constituent part of a combination product. Such an HCT/P is subject to part 1271 of this chapter and is also regulated as a drug, device, and/or biological product.
[FDA 21CFR4]
human cell, tissue, or cellular or tissue-based product (HCT/P) regulated as a devicean HCT/P as defined in 1271.3(d) of this chapter that does not meet the criteria in 1271.10(a) and that is also regulated as a device
[FDA 21CFR801, FDA 21CFR803, FDA 21CFR806, FDA 21CFR810, FDA 21CFR814, FDA 21CFR820, FDA 21CFR821, FDA 21CFR822, FDA 21CFR830]
human subjectan individual who is or becomes a participant in research, either as a recipient of the test article or as a control. A subject may be either a healthy human or a patient.
[FDA 21CFR50, FDA 21CFR56]
humanitarian device exemption (HDE)a premarket approval application submitted pursuant to this subpart seeking a humanitarian device exemption from the effectiveness requirements of sections 514 and 515 of the act as authorized by section 520(m)(2) of the act
[FDA 21CFR814]
Note: An HDE is a marketing application for an HUD.
humanitarian use device (HUD)a medical device intended to benefit patients in the treatment or diagnosis of a disease or condition that affects or is manifested in not more than 8,000 individuals in the United States per year
[FDA 21CFR814]
implanta device that is placed into a surgically or naturally formed cavity of the human body if it is intended to remain there for a period of 30 days or more. FDA may, in order to protect public health, determine that devices placed in subjects for shorter periods are also “implants” for purposes of this part.
[FDA 21CFR812]
implantable devicea device that is intended to be placed in a surgically or naturally formed cavity of the human body. A device is regarded as an implantable device for the purpose of this part only if it is intended to remain implanted continuously for a period of 30 days or more, unless the Commissioner of Food and Drugs determines otherwise in order to protect human health
[FDA 21CFR801]
implantable deviceany device, including those that are partially or wholly absorbed, which is intended:
— to be totally introduced into the human body, or
— to replace an epithelial surface or the surface of the eye, by clinical intervention and which is intended to remain in place after the procedure
Note: Any device intended to be partially introduced into the human body by clinical intervention and intended to remain in place after the procedure for at least 30 days shall also be deemed to be an implantable device
[EU MDR 2017/745]
implantable medical devicemedical device which can only be removed by medical or surgical intervention and which is intended to
- be totally or partially introduced into the human body or a natural orifice, or
- replace an epithelial surface or the surface of the eye, and
- remain after the procedure for at least 30 days
Note: This definition of implantable medical device includes active implantable medical device
[ISO 13485:2016]
in vitro diagnostic medical deviceany medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used
in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally for the purpose of providing information on one or more of the following:
(a) concerning a physiological or pathological process or state;
(b) concerning congenital physical or mental impairments;
(c) concerning the predisposition to a medical condition or a disease;
(d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions;
(f) to define or monitoring therapeutic measures.
Note: Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices
[EU IVDR 2017/746]
in vitro diagnostic medical device (IVD medical device)medical device, whether used alone or in combination, intended by the manufacturer for the
in vitro examination of specimens derived from the human body solely or principally to provide information for diagnostic, monitoring, or compatibility purposes
Note: IVD medical devices include reagents, calibrators, control materials, specimen receptacles, software and related instruments or apparatus or other articles and are used, for example, for the following test purposes: diagnosis, aid to diagnosis, screening, monitoring, predisposition, prognosis, prediction, determination of physiological status.
[ISO 20916:2019]
In vitro diagnostic productsthose reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body. These products are devices as defined in section 201(h) of the Federal Food, Drug, and Cosmetic Act (the act), and may also be biological products subject to section 351 of the Public Health Service Act.
[FDA 21CFR809]
incidentany malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any undesirable side-effect
[EU MDR 2017/745]
incidentany malfunction or deterioration in the characteristics or performance of a device made available on the market, including use-error due to ergonomic features, as well as any inadequacy in the information supplied by the manufacturer and any harm as a consequence of a medical decision, action taken or not taken on the basis of information or result(s) provided by the device
[EU IVDR 2017/746]
independent reviewera qualified person who does not have direct responsibility for the design work being reviewed that participates in the design review and provides oversight for design control content
indications for usea general description of the disease or condition the device will diagnose, treat, prevent, cure, or mitigate, including a description of the patient population for which the device is intended.
[FDA 21CFR814, FDA guidance: Deciding When to Submit...(2014)]
initial importerAny importer who furthers the marketing of a device from a foreign manufacturer to the person who makes final delivery or sale of the device to the ultimate consumer or user, but does not repackage, or otherwise change the container, wrapper, or labeling of the device or device package. The initial importer must have a physical address in the United States staffed by individuals responsible for ensuring the compliance of imported devices with all applicable FDA laws and regulations.
[FDA website: Who Must Register...]
inspectiondetermination of conformity to specified requirements
Note 1: If the result of an inspection shows conformity, it can be used for purposes of verification.
Note 2: The result of an inspection can show conformity or nonconformity or a degree of conformity.
[ISO 9000:2015]
inspection and testing processprocess of combining various inputs to verify that the product meets its product design specifications
installation controlsmeasures that ensure that the medical device is installed at the customer location such that product design specifications are met with identification and traceability maintained
installation processprocess of combining various inputs in order to install a medical device such that the medical device is ready for use
institutional review board (IRB)any board, committee, or other group formally designated by an institution to review, to approve the initiation of, and to conduct periodic review of, biomedical research involving human subjects. The primary purpose of such review is to assure the protection of the rights and welfare of the human subjects. The term has the same meaning as the phrase institutional review committee as used in section 520(g) of the act.
[FDA 21CFR56]
institutional review board (IRB)any board, committee, or other group formally designated by an institution to review biomedical research involving humans as subjects, to approve the initiation of and conduct periodic review of such research. The term has the same meaning as the phrase institutional review committee as used in section 520(g) of the act.
[FDA 21CFR50]
institutional review board (IRB)any board, committee, or other group formally designated by an institution to review biomedical research involving subjects and established, operated, and functioning in conformance with part 56. The term has the same meaning as “institutional review committee” in section 520(g) of the act.
[FDA 21CFR812]
instructions for usethe information provided by the manufacturer to inform the user of a device's intended purpose and proper use and of any precautions to be taken
[EU MDR 2017/745, EU IVDR 2017/746]
instructions for use (IFU)the information provided by the manufacturer to inform the user of a device's intended purpose and proper use and of any precautions to be taken
[EU MDR 2017/745, EU IVDR 2017/746]
instructions for use (IFU) (directions for use)those parts of accompanying documents giving the necessary information for safe and proper use and operation of the equipment
[IEC TR 60788:2004]
intended purposethe use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation
[EU MDR 2017/745]
intended purposethe use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements or as specified by the manufacturer in the performance evaluation
[EU IVDR 2017/746]
intended usethe general purpose of the device or its function. The intended use of a device encompasses the indications for use.
[FDA guidance: The 510(k) Program...(2014), FDA guidance: Deciding When to Submit...(2014)]
intended useobjective intent of the manufacturer regarding the use of a product, process or service as reflected in the specifications, instructions and information provided by the IVD manufacturer
Note: Intended use statements for IVD labelling can include two components: a description of the functionality of the IVD medical device (e.g. an immunochemical measurement procedure for the detection of analyte “x” in serum or plasma), and a statement of the intended medical use of the examination results.
[ISO 20916:2019]
intended use (intended purpose)use for which a product, process, or service is intended according to the specifications, instructions and information provided by the manufacturer
Note 1: The intended medical indication, patient population, part of the body or type of tissue interacted with, user profile, use environment, and operating principle are typical elements of the intended use.
[ISO 14971:2019]
Note 2: Intended use should not be confused with normal use. While both include the concept of use as intended by the manufacturer, intended use focuses on the medical purpose while normal use incorporates not only the medical purpose, but maintenance, transport, etc. as well.
[IEC 60601-1:2012]
intended usesthe words intended uses or words of similar import in §§801.5, 801.119, and 801.122 refer to the objective intent of the persons legally responsible for the labeling of devices. The intent is determined by such persons' expressions or may be shown by the circumstances surrounding the distribution of the article. This objective intent may, for example, be shown by labeling claims, advertising matter, or oral or written statements by such persons or their representatives. It may be shown by the circumstances that the article is, with the knowledge of such persons or their representatives, offered and used for a purpose for which it is neither labeled nor advertised.
Note: The intended uses of an article may change after it has been introduced into interstate commerce by its manufacturer. If, for example, a packer, distributor, or seller intends an article for different uses than those intended by the person from whom he received the devices, such packer, distributor, or seller is required to supply adequate labeling in accordance with the new intended uses. But if a manufacturer knows, or has knowledge of facts that would give him notice that a device introduced into interstate commerce by him is to be used for conditions, purposes, or uses other than the ones for which he offers it, he is required to provide adequate labeling for such a device which accords with such other uses to which the article is to be put.
[FDA 21CFR801]
investigational devicea device, including a transitional device, that is the object of an investigation
[FDA 21CFR812]
investigational devicea device that is assessed in a clinical investigation
[EU MDR 2017/745]
investigational device exemption (IDE)an approved or considered approved investigational device exemption under section 520(g) of the act and parts 812 and 813
[FDA 21CFR814]
investigational medical devicemedical device being assessed for clinical performance, effectiveness, or safety in a clinical investigation
Note 1: This includes medical devices already on the market that are being evaluated for new intended uses, new populations, new materials or design changes.
Note 2: This includes medical devices already on the market that are being evaluated within their intended use in a post-market clinical investigation (interventional or non-interventional).
Note 3: For the purpose of this document, the terms “investigational medical device” and “investigational device” are used interchangeably.
[ISO 14155:2020]
itemany part, component, device, subsystem, functional unit, equipment or system that can be individually considered
[IEC 60812:2006]
labelany display of written, printed, or graphic matter on the immediate container of any article, or any such matter affixed to any consumer commodity or affixed to or appearing upon a package containing any consumer commodity
[FDA 21CFR1]
labela display of written, printed, or graphic matter upon the immediate container of any article; and a requirement made by or under authority of this chapter that any word, statement, or other information appear on the label shall not be considered to be complied with unless such word, statement, or other information also appears on the outside container or wrapper, if any there be, of the retail package of such article, or is easily legible through the outside container or wrapper.
Note: The term "immediate container" does not include package liners.
[OLRC 21USC9]
labelthe written, printed or graphic information appearing either on the device itself, or on the packaging of each unit or on the packaging of multiple devices
[EU MDR 2017/745]
labelwritten, printed or graphic information provided upon the medical device itself
[ISO 15223-1:2016]
labelingall written, printed, or graphic matter accompanying an article at any time while such article is in interstate commerce or held for sale after shipment or delivery in interstate commerce
Note: The term 'accompanying' is interpreted liberally to mean more than physical association with the product. It extends to posters, tags, pamphlets, circulars, booklets, brochures, instruction books, direction sheets, fillers, etc. 'Accompanying' also includes labeling that is brought together with the device after shipment or delivery for shipment in interstate commerce.
[FDA 21CFR1]
labelingall labels and other written, printed, or graphic matter (1) upon any article or any of its containers or wrappers, or (2) accompanying such article.
[OLRC 21USC9]
labellinglabel, instructions for use, and any other information that is related to identification, technical description, intended purpose and proper use of the medical device, but excluding shipping documents
[ISO 13485:2016]
lay personan individual who does not have formal education in a relevant field of healthcare or medical discipline
[EU MDR 2017/745, EU IVDR 2017/746]
life-cycleall phases in the life of a medical device, from the initial conception to final decommissioning and disposal
[ISO 13485:2016, ISO 14971:2019]
maintenance (routine servicing)combination of all technical and associated administrative actions intended to retain an item at/or restore it to a state in which it can perform its required function
[ISO 17665-1:2006]
Note: Routine servicing is any regularly scheduled maintenance of a device, including the replacement of parts at the end of their normal life expectancy, e.g., calibration, replacement of batteries, and responses to normal wear and tear. Repairs of an unexpected nature, replacement of parts earlier than their normal life expectancy, or identical repairs or replacements of multiple units of a device are not routine servicing.
[FDA 21CFR806]
manufacturernatural or legal person with responsibility for design and/or manufacture of a medical device with the intention of making the medical device available for use, under his name; whether or not such a medical device is designed and/or manufactured by that person himself or on his behalf by another person(s)
Note 1: This "natural or legal person" has ultimate legal responsibility for ensuring compliance with all applicable regulatory requirements for the medical devices in the countries or jurisdictions where it is intended to be made available or sold, unless this responsibility is specifically imposed on another person by the Regulatory Authority (RA) within that jurisdiction.
Note 2: The manufacturer’s responsibilities are described in other GHTF guidance documents. These responsibilities include meeting both pre-market requirements and post-market requirements, such as adverse event reporting and notification of corrective actions.
Note 3: "Design and/or manufacture", as referred to in the above definition, may include specification development, production, fabrication, assembly, processing, packaging, repackaging, labelling, relabelling, sterilization, installation, or remanufacturing of a medical device; or putting a collection of devices, and possibly other products, together for a medical purpose.
Note 4: Any person who assembles or adapts a medical device that has already been supplied by another person for an individual patient, in accordance with the instructions for use, is not the manufacturer, provided the assembly or adaptation does not change the intended use of the medical device.
Note 5: Any person who changes the intended use of, or modifies, a medical device without acting on behalf of the original manufacturer and who makes it available for use under his own name, should be considered the manufacturer of the modified medical device.
Note 6: An authorized representative, distributor or importer who only adds its own address and contact details to the medical device or the packaging, without covering or changing the existing labelling, is not considered a manufacturer.
Note 7: To the extent that an accessory is subject to the regulatory requirements of a medical device, the person responsible for the design and/or manufacture of that accessory is considered to be a manufacturer.
[ISO 13485:2016, ISO 14971:2019]
manufacturerany person who designs, manufactures, fabricates, assembles, or processes a finished device. Manufacturer includes but is not limited to those who perform the functions of contract sterilization, installation, relabeling, remanufacturing, repacking, or specification development, and initial distributors of foreign entities performing these functions
[FDA 21CFR820]
manufacturera natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark
[EU MDR 2017/745, EU IVDR 2017/746]
manufacturerany person who manufactures, prepares, propagates, compounds, assembles, or processes a device by chemical, physical, biological, or other procedure. The term includes any person who either:
(1) Repackages or otherwise changes the container, wrapper, or labeling of a device in furtherance of the distribution of the device from the original place of manufacture;
(2) Initiates specifications for devices that are manufactured by a second party for subsequent distribution by the person initiating the specifications;
(3) Manufactures components or accessories that are devices that are ready to be used and are intended to be commercially distributed and intended to be used as is, or are processed by a licensed practitioner or other qualified person to meet the needs of a particular patient; or
(4) Is the U.S. agent of a foreign manufacturer.
[FDA 21CFR803]
manufacturer[organization that] makes by chemical, physical, biological, or other procedures, any article that meets the definition of "device" in Section 201(h) of the Federal Food, Drug, and Cosmetic (FD&C) Act
[FDA website: Who Must Register...]
manufacturing materialany material or substance used in or used to facilitate the manufacturing process, a concomitant constituent, or a byproduct constituent produced during the manufacturing process, which is present in or on the finished device as a residue or impurity not by design or intent of the manufacturer
[FDA 21CFR820]
market withdrawala correction or removal of a distributed device that involves a minor violation of the act that would not be subject to legal action by FDA or that involves no violation of the act, e.g., normal stock rotation practices
[FDA 21CFR806]
materialsynthetic or natural polymer, metal or alloy, ceramic, or composite, including tissue rendered non-viable, used as a
medical device or any part thereof
[ISO 10993-1:2018]
medical deviceinstrument, apparatus, implement, machine, appliance, implant, reagent for in vitro use, software, material or other similar or related article, intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the specific medical purpose(s) of:
- diagnosis, prevention, monitoring, treatment or alleviation of disease;
- diagnosis, monitoring, treatment, alleviation of or compensation for an injury;
- investigation, replacement, modification, or support of the anatomy or of a physiological process;
- supporting or sustaining life;
- control of conception,
- disinfection of medical devices;
- providing information by means of in vitro examination of specimens derived from the human body;
and does not achieve its primary intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its intended function by such means
Note 1: Products which may be considered to be medical devices in some jurisdictions but not in others include:
- disinfection substances;
- aids for persons with disabilities;
- devices incorporating animal and/or human tissues;
- devices for in vitro fertilization or assisted reproduction technologies.
[ISO 13485:2016]
Note 2: Products which may be considered to be medical devices in some jurisdictions but not in others include:
— items specifically intended for cleaning or sterilization of medical devices;
— pouches, reel goods, sterilization wrap and reusable containers for packaging of medical devices for sterilization.
[ISO 11139:2018]
Note 3: Medical devices include dental devices.
[ISO 10993-1:2018]
medical deviceany instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes: — diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease, — diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability, — investigation, replacement or modification of the anatomy or of a physiological or pathological process or state, — providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations, and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
Note: The following products shall also be deemed to be medical devices:
— devices for the control or support of conception;
— products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.
[EU MDR 2017/745]
medical device (device)an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is—
(1) recognized in the official National Formulary, or the United States Pharmacopeia, or any supplement to them,
(2) intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
(3) intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes. The term "device" does not include software functions excluded pursuant to section 360j(o) of this title.
[OLRC 21USC9]
medical device file (MDF)file containing or referencing documents generated to demonstrate conformity to the requirement of this International Standard and compliance with applicable regulatory requirements
Note: The content of the file(s) shall include, but not limited to:
a) General description of the medical device, intended use/purpose, and labelling, including any instructions for use
b) specifications for product;
c) specifications or procedures for manufacturing, packaging, storage, handling and distribution;
d) procedures for measuring and monitoring;
e) as appropriate, requirements for installation;
f) as appropriate, procedures for servicing.
[ISO 13485:2016 derived]
medical device report (MDR)report submitted to FDA of suspected device-associated deaths, serious injuries and/or malfunctions
[FDA website: Medical Device Reporting (MDR)...]
medical device softwaresoftware system that has been developed for the purpose of being incorporated into the medical device being developed or that is intended for use as a medical device
Note: This includes a medical device software product, which then is a medical device in its own right.
[IEC 62304:2015]
medical systemsystem containing at least one medical device
nonconformity (nonconformance, NC)nonfulfillment of a specified requirement
[FDA 21CFR820]
nonconformity (nonconformance, NC)non-fulfilment of a requirement
[ISO 9000:2015]
normal useoperation, including routine inspection and adjustments by any user, and stand-by, according to the instructions for use or in accordance with generally accepted practice for those medical devices provided without instructions for use
Note 1: Normal use should not be confused with intended use. While both include the concept of use as intended by the manufacturer, intended use focuses on the medical purpose while normal use incorporates not only the medical purpose, but maintenance, transport, etc. as well.
Note 2: Use error can occur in normal use.
Note 3: Medical devices that can be used safely without instructions for use are exempted from having instructions for use by some authorities with jurisdiction.
Note 4: The figure below shows the relationships of the types of use.
[IEC 62366-1:2015]
notified body (NB)a conformity assessment body designated in accordance with this Regulation
[EU MDR 2017/745, EU IVDR 2017/746]
OEM producta product purchased from an original equipment manufacturer with the intention of being made available on the market by the purchasing organization which does not own the product design
Note: In this scenario, the original equipment manufacturer owns the product design and is considered a supplier to the purchasing organization that makes the product available on the market.
off-label useuse of a health product in a manner that is not approved by the regulatory authority with jurisdiction at the location of use
operating principleThe mode of operation or mechanism of action through which a device fulfills (or achieves) its intended use
Note: An example of a change in operating principle would be using a new algorithm to compress images in a picture archiving and communications system. For an IVD, an example would be a change from immunofluorescence to ELISA.
[FDA guidance: Deciding When to Submit...(2014)]
outputresult of a process
Note: Whether an output of the organization is a product or a service depends on the preponderance of the characteristics involved, e.g. a painting for sale in a gallery is a product whereas supply of a commissioned painting is a service, a hamburger bought in a retail store is a product whereas receiving an order and serving a hamburger ordered in a restaurant is part of a service.
[ISO 9000:2015]
overall residual risk (ORR)combination of all individual residual risks
overheadthe ongoing business expenses not directly attributed to creating a product or service
Note: Overhead is important for budgeting purposes but also for determining how much a company must charge for its products or services to make a profit. In short, overhead is any expense incurred to support the business while not being directly related to a specific product or service.
[Investopedia]
packagingany wrapping, containers, etc., used to protect, to preserve the sterility of, or to group medical devices.
[FDA guidance: Deciding When to Submit...(2014)]
patentthe grant of a property right to the inventor of an invention
[USPTO]
patientliving being (person) undergoing a medical, surgical or dental procedure
Note: A patient can be a user.
[IEC 62366-1:2015]
patient-specific devices (patient-matched devices, customized devices)Patient-specific devices are, in general, ones in which ranges of different specifications have been approved or cleared to treat patient populations that can be studied clinically. Premarket submissions for such devices are sometimes referred to as “envelope” submissions because their approval or clearance covers the entire range of specifications data they contain to support. The final manufacturing of these devices can be delayed until physicians provide imaging data or other information to the manufacturer to finalize device specifications within cleared or approved ranges. As a result, such devices are specifically tailored to patients. For example, a manufacturer of an ankle replacement device could submit a 510(k) to cover a range of specifications for different system components to accommodate multiple patients with different anatomical characteristics.
Note: While some in industry have sometimes colloquially referred to these devices as “customized,” they are not custom devices meeting the FD&C Act custom device exemption requirements unless they comply with all of the criteria of section 520(b). Marketing applications are required for these device types because the devices and patient populations can be defined and studied.
[FDA guidance: Custom Device Exemption (2014)]
peer reviewa formal (documented) or informal review focused on a specific aspect (topic) of product development
Note: Peer reviews are typically narrower in scope than design reviews and are considered verification activities that precede and/or facilitate the successful completion of design reviews. Other names for peer reviews include: functional review, cross-functional review, technical review, topic review, etc.
performance evaluationassessment and analysis of data to establish or verify the ability of an
in vitro diagnostic medical device to achieve its intended use
[ISO 13485:2016]
performance evaluationan assessment and analysis of data to establish or verify the scientific validity, the analytical and, where applicable, the clinical performance of a device
[EU IVDR 2017/746]
performance studystudy undertaken to establish or confirm the analytical or clinical performance of a device
[EU IVDR 2017/746]
phase reviewa documented review of product development life-cycle phase activities to confirm process function alignment, completion of required tasks/deliverables, and leadership buy-in to complete the phase
placing on the marketthe first making available of a device, other than an investigational device, on the Union market
[EU MDR 2017/745]
placing on the marketthe first making available of a device, other than a device for performance study, on the Union market
[EU IVDR 2017/746]
post-market (post-launch)after placement on the market
[ISO 13485:2016]
post-market surveillance (PMS)systematic process to collect and analyze experience gained from medical devices that have been placed on the market
[ISO 13485:2016]
post-market surveillance (PMS)all activities carried out by manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market, make available on the market or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions
[EU MDR 2017/745, EU IVDR 2017/746]
post-productionpart of the life-cycle of the medical device after the design has been completed and the medical device has been manufactured
Examples: Transportation, storage, installation, product use, maintenance, repair, product changes, decommissioning and disposal.
[ISO 14971:2019]
postmarket surveillance (PMS)the active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device
[FDA 21CFR822]
pre-market (pre-launch)before placement on the market
preamendments devicea device commercially distributed in the United States prior to May 28, 1976 that has not been significantly changed or modified since then, and for which premarket approval has not been required under section 515(b) of the FD&C Act
[FDA guidance: Deciding When to Submit...(2014)]
precautions, warnings, and contraindications
- Precautions describe any special care to be exercised by a practitioner or patient for the safe and effective use of a device. This definition also includes limitations stated for IVDs.
- Warnings describe serious adverse reactions and potential safety hazards that can occur in the proper use or misuse of a device, along with consequent limitations in use and mitigating steps to take if they occur.
- Contraindications describe situations in which the device should not be used because the risk of use clearly outweighs any reasonably foreseeable benefits.
[FDA guidance: Deciding When to Submit...(2014)]
predicate devicea legally marketed device (as defined in 21 CFR 807.92(a)(3)) to which a new device may be compared for a determination regarding substantial equivalence because the devices have the same intended use and the same technological characteristics or different technological characteristics that do not raise different questions of safety and effectiveness
[FDA guidance: The 510(k) Program...(2014)]
premarket approval application (PMA)any premarket approval application for a class III medical device, including all information submitted with or incorporated by reference therein. “PMA” includes a new drug application for a device under section 520(1) of the act.
[FDA 21CFR814]
preventive actionaction to eliminate the cause of a potential nonconformity or other potential undesirable situation
[ISO 9000:2015]
procedurespecified way to carry out an activity or a process
[ISO 9000:2015, ISO 14971:2019]
processset of interrelated or interacting activities that use inputs to deliver an intended result
Note 1: Whether the “intended result” of a process is called output, product or service depends on the context of the reference.
Note 2: Inputs to a process are generally the outputs of other processes and outputs of a process are generally the inputs to other processes.
Note 3: Two or more interrelated and interacting processes in series can also be referred to as a process.
Note 4: Processes in an organization are generally planned and carried out under controlled conditions to add value.
Note 5: A process where the conformity of the resulting output cannot be readily or economically validated is frequently referred to as a “special process”.
[ISO 9000:2015, ISO 14971:2019]
process validationestablishing by objective evidence that a process consistently produces a result or product meeting its predetermined specifications
[FDA 21CFR820]
processingCleaning, disinfection, or sterilization of an unused medical device
productcomponents, manufacturing materials, in-process devices, finished devices, and returned devices
[FDA 21CFR820]
productresult of a process
Note 1: There are four generic product categories, as follows:
- services (e.g. transport);
- software (e.g. computer programs, dictionary);
- hardware (e.g. engine mechanical part);
- processed materials (e.g. lubricant).
Many products comprise elements belonging to different generic product categories. Whether the product is then called service, software, hardware or processed material depends on the dominant element. For example, the offered product "automobile" consists of hardware (e.g. tyres), processed materials (e.g. fuel, colling liquid), software (e.g. engine control software, driver's manual), and service (e.g. operating explanations given by the salesman).
Note 2: Service is the result of at least one activity necessarily performed at the interface between the supplier and the customer and is generally intangible. Provision of a service can involve, for example, the following:
- An activity performed on a customer-supplied tangible prodict (e.g. automobile to be repaired);
- an activity performed on a customer-supplied intangible product (e.g. the income statement needed to prepare a tax return);
- the delivery of an intangible product (e.g. the delivery of information in the context of knowledge transmission);
- the creation of ambience for the customer (e.g. in hotels and restaurants).
Software consists of information and is generally intangible and can be in the form of approaches, transactions or procedures.
Hardware is generally tangible and its amount is a countable characteristic. Processed materials are generally tangible and their amount is a continuous characteristic. Hardware and processed materials often are referred to as goods.
Note 3: This definition of "product" differs from the definition given in ISO 9000:2015.
[ISO 13485:2016]
Product Development Protocol (PDP)a submission as set forth in section 515(f) of the Federal Food, Drug, and Cosmetic Act
[FDA 21CFR4]
product failure modefailure of the medical device or medical device accessory (including labeling and packaging) to perform or function as intended including any deviations from the device's performance specifications
product familygroup or subgroup of product characterized by similar attributes determined to be equivalent for evaluation and processing purposes
[ISO 11139:2018]
product field action (PFA)a firm's removal or correction of a marketed product
Note: PFA includes recalls and market withdrawals, but not stock recoveries.
production controlsmeasures that ensure that the produced product (at the manufacturer’s location) conforms to product design specifications with identification and traceability maintained throughout production
production processprocess of combining various inputs in order to produce a packaged medical device that is suitable for delivery
profitthe financial benefit realized when revenue generated from a business activity exceeds the expenses, costs, and taxes involved in sustaining the activity in question
Note: Profit is calculated as total revenue less total expenses. Any profits earned funnel back to business owners, who choose to either pocket the cash or reinvest it back into the business.
[Investopedia]
profit and loss statement (P&L)a financial statement that summarizes the revenues, costs, and expenses incurred during a specified period, usually a fiscal quarter or year
Note: The P&L statement provides information about a company's ability or inability to generate profit by increasing revenue, reducing costs, or both. The P&L statement is also known as the statement of profit and loss, income statement, statement of operations, statement of financial results or income, earnings statement, or expense statement.
[Investopedia]
projectunique process, consisting of a set of coordinated and controlled activities with start and finish dates, undertaken to achieve an objective conforming to specific requirements, including the constraints of time, cost and resources
Note 1: An individual project can form part of a larger project structure and generally has a defined start and finish date.
Note 2: In some projects the objectives and scope are updated and the product or service characteristics defined progressively as the project proceeds.
Note 3: The output of a project can be one or several units of product or service.
Note 4: The project’s organization is normally temporary and established for the lifetime of the project.
Note 5: The complexity of the interactions among project activities is not necessarily related to the project size.
[ISO 9000:2015]
project managementplanning, organizing, monitoring, controlling and reporting of all aspects of a project, and the motivation of all those involved in it to achieve the project objectives
[ISO 9000:2015]
project teampersonnel with direct responsibility for the completion of product development project tasks and/or deliverables
project team meetingmeeting to discuss project progress, key project decisions, and resolve any action items (open issues)
prospective surveillancethe subjects are identified at the beginning of the surveillance and data or other information will be collected from that time forward (as opposed to retrospective surveillance)
[FDA 21CFR822]
purchased productproduct provided by a party outside the organization's quality management system
Note: The provision of product does not necessarily infer a commercial or financial arrangement
[ISO 13485:2016]
purchasing controlsmeasures that ensure that all purchased or otherwise received products and services conform to specified purchasing requirements
putting into servicethe stage at which a device, other than an investigational device, has been made available to the final user as being ready for use on the Union market for the first time for its intended purpose
[EU MDR 2017/745]
putting into servicethe stage at which a device, other than a device for performance study, has been made available to the final user as being ready for use on the Union market for the first time for its intended purpose
[EU IVDR 2017/746]
qualificationactivities undertaken to demonstrate that utilities, equipment, and methods or modes are suitable for their intended use and perform properly
Note: Qualification of equipment and/or processes generally includes installation qualification, operational qualification, and performance qualification.
[ISO 11139:2018]
qualificationdocumented process used by the health care product manufacturer to assure the reliability and capability of equipment and/or processes before approval for use in manufacturing
Note: Qualification of equipment and/or processes generally includes installation qualification, operational qualification and performance qualification.
[ISO 13408-1:2015]
qualitythe totality of features and characteristics that bear on the ability of a device to satisfy fitness-for-use, including safety and performance
[FDA 21CFR820]
qualitydegree to which a set of inherent characteristics of an object fulfils requirements
Note 1: The term “quality” can be used with adjectives such as poor, good or excellent.
Note 2: “Inherent”, as opposed to “assigned”, means existing in the object
[ISO 9000:2015]
quality management system (QMS)part of a management system with regard to quality
[ISO 9000:2015]
quality system (QS)the organizational structure, responsibilities, procedures, processes, and resources for implementing quality management
[FDA 21CFR820]
reasonably foreseeable misuseuse of a product or system in a way not intended by the manufacturer, but which can result from readily predictable human behaviour
Note 1: Readily predictable human behaviour includes the behaviour of all types of users, e.g. lay and professional users.
Note 2: Reasonably foreseeable misuse can be intentional or unintentional.
[ISO 14971:2019]
recallany measure aimed at achieving the return of a device that has already been made available to the end user
[EU MDR 2017/745, EU IVDR 2017/746]
recalla firm's removal or correction of a marketed product that the Food and Drug Administration considers to be in violation of the laws it administers and against which the agency would initiate legal action, e.g., seizure. Recall does not include a market withdrawal or a stock recovery.
[FDA 21CFR7]
recorddocument stating results achieved or providing evidence of activities performed
[ISO 9000:2015, ISO 14971:2019]
regulatory authority (authority having jurisdiction)government agency or other entity that exercises a legal right to control the use or sale of medical devices within its jurisdiction, and can take legal action to ensure that medical devices marketed within its jurisdiction comply with legal requirements
[ISO 20916:2019]
regulatory requirementobligatory requirement specified by an authority mandated by a legislative body
[ISO 9000:2015]
relabeler[organization that] changes the content of the labeling from that supplied from the original manufacturer for distribution under the establishment's own name. A relabeler does not include establishments that do not change the original labeling but merely add their own name.
[FDA website: Who Must Register...]
remanufacturerany person who processes, conditions, renovates, repackages, restores, or does any other act to a finished device that significantly changes the finished device's performance or safety specifications, or intended use
[FDA 21CFR820, FDA website: Who Must Register...]
repackager[organization that] packages finished devices from bulk or repackages devices made by a manufacturer into different containers (excluding shipping containers)
[FDA website: Who Must Register...]
repairaction on a nonconforming product or service to make it acceptable for the intended use
Note 1: A successful repair of a nonconforming product or service does not necessarily make the product or service conform to the requirements. It can be that in conjunction with a repair a concession is required.
Note 2: Repair includes remedial action taken on a previously conforming product or service to restore it for use, for example as part of maintenance.
Note: Repair can affect or change parts of the nonconforming product or service.
[ISO 9000:2015 ]
repeatabilitycondition of measurement, out of a set of conditions that includes the same measurement procedure, same operators, same measuring system, same operating conditions and same location, and replicate measurements on the same or similar objects over a short period of time
[ISO 11139:2018]
repeatabilityA measure of how well one can obtain the same observed value when measuring the same part or sample over and over using the same measuring device
Note: Repeatability is sometimes referred to as equipment variation, or EV.
[Crossley, 2000]
reprocessed single-use devicean original device that has previously been used on a patient and has been subjected to additional processing and manufacturing for the purpose of an additional single use on a patient. The subsequent processing and manufacture of a reprocessed single-use device shall result in a device that is reprocessed within the meaning of this definition.
Note 1: A single-use device that meets the definition under clause (A) shall be considered a reprocessed device without regard to any description of the device used by the manufacturer of the device or other persons, including a description that uses the term "recycled" rather than the term "reprocessed".
Note 2: The term "original device" means a new, unused single-use device.
Note 3: The term "critical reprocessed single-use device" means a reprocessed single-use device that is intended to contact normally sterile tissue or body spaces during use.
Note 4: The term "semi-critical reprocessed single-use device" means a reprocessed single-use device that is intended to contact intact mucous membranes and not penetrate normally sterile areas of the body.
[OLRC 21USC9]
reprocessingvalidated processes used to render a medical device, which has been previously used or contaminated, fit for a subsequent single use. These processes are designed to remove soil and contaminants by cleaning and to inactivate microorganisms by disinfection or sterilization
[FDA guidance: Reprocessing Medical Devices...(2017), FDA guidance: Deciding When to Submit...(2014)]
reprocessinga process carried out on a used device in order to allow its safe reuse including cleaning, disinfection, sterilisation and related procedures, as well as testing and restoring the technical and functional safety of the used device
[EU MDR 2017/745]
reprocessor of single-use device[organization that] performs remanufacturing operations on a single-use device
[FDA website: Who Must Register...]
reproducibilityHow well others agree with our measurement when measuring the same sample using the same measuring device
Note: Reproducibility is sometimes referred to as appraiser variation, or AV.
[Crossley, 2000]
reproducibilitycondition of measurement, out of a set of conditions that includes different locations, processors, measuring systems, and replicate measurements on the same or similar objects
[ISO 11139:2018]
residual riskrisk remaining after risk control measures have been taken
[ISO 14971:2019]
reusable medical devicea device intended for repeated use either on the same or different patients, with appropriate cleaning and other reprocessing between uses
[FDA guidance: Deciding When to Submit...(2014), FDA guidance: Reprocessing Medical Devices...(2017)]
reusable medical devicemedical device designated or intended by the manufacturer as suitable for processing and reuse
[ISO 11139:2018]
revenueincome generated from normal business operations including discounts and deductions for returned merchandise
Note: Revenue is the top line or gross income figure from which costs are subtracted to determine net income.
[Investopedia]
reworkaction taken on a nonconforming product so that it will fulfill the specified DMR requirements before it is released for distribution
[FDA 21CFR820]
reworkaction on a nonconforming product or service to make it conform to the requirements
[ISO 9000:2015]
riskcombination of the probability of occurrence of harm and the severity of that harm
[ISO 14971:2019, ISO 13485:2016]
riskeffect of uncertainty on objectives
Note 1: An effect is a deviation from the expected. It can be positive, negative or both, and can address, create or result in opportunities and threats.
Note 2: Objectives can have different aspects and categories, and can be applied at different levels.
Note 3: Risk is usually expressed in terms of risk sources, potential events, their consequences and their likelihood.
[ISO 31000:2018]
risk analysissystematic use of available information to identify hazards and to estimate the risk
[ISO 14971:2019]
risk assessmentoverall process comprising a risk analysis and a risk evaluation
[ISO 14971:2019]
risk controlprocess in which decisions are made and measures implemented by which risks are reduced to, or maintained within, specified levels
[ISO 14971:2019]
risk control measure (RCM)measure implemented by which a risk is reduced to, or maintained within, a specified level
Note: Risk control measures can be divided into two categories:
1. Existing risk control measures - risk control measures that already exist within the design (product and operations processes)
2. Additional risk control measures - new risk control measures identified during risk control option analysis
risk estimationprocess used to assign values to the probability of occurrence of harm and the severity of that harm
[ISO 14971:2019]
risk evaluationprocess of comparing the estimated risk against given risk criteria to determine the acceptability of the risk
[ISO 14971:2019]
risk managementsystematic application of management policies, procedures and practices to the tasks of analysing, evaluating, controlling and monitoring risk
[ISO 14971:2019, ISO 13485:2016]
risk management board (RMB)designated group of subject matter experts and managers who have responsibility for the supervision of all risk management activities at a manufacturer including authorization to balance benefits against residual risks to make final decisions regarding the acceptability of overall residual risk
risk management file (RMF)set of records and other documents that are produced by risk management
[ISO 14971:2019]
risk management plan (RMP)overall plan for risk management activities for the particular medical device under consideration
risk management report (RMR)a summary of the review of the final results of the risk management process for the particular medical device under consideration
risk management teampersons performing risk management tasks
risk matrixmatrix that graphically displays risk regions and risk levels
risk parallaxinstance where one risk has been allowed to increase in order that another risk could be reduced
Example: The risk to one person (the user) is allowed to increase so that the risk to another (the patient) can be reduced.
[ISO/TR 24971:2013 derived]
risk tablemain risk management summary table for documenting risk analysis, risk evaluation, and risk control activities
routine servicingany regularly scheduled maintenance of a device, including the replacement of parts at the end of their normal life expectancy, e.g., calibration, replacement of batteries, and responses to normal wear and tear. Repairs of an unexpected nature, replacement of parts earlier than their normal life expectancy, or identical repairs or replacements of multiple units of a device are not routine servicing
[FDA 21CFR806]
safetyfreedom from unacceptable risk
[ISO 14971:2019]
Note: Safety can be divided into two parts:
1. Clinical safety - safety from clinical complications
2. Technical safety - safety from medical device hazards
scrapaction on a nonconforming product or service to preclude its originally intended use
Example: Recycling, destruction.
Note: In a nonconforming service situation, use is precluded by discontinuing the service.
[ISO 9000:2015]
securityprotection of information and data so that unauthorized persons or systems cannot read or modify them and authorized persons or systems are not denied access to them
[IEC 62304:2015]
securitya condition that results from the establishment and maintenance of protective measures that ensure a state of inviolability from hostile acts or influences, where hostile acts or influences could be intentional or unintentional
[ISO/TR 24971:2020]
serious injuryinjury or illness that:
a) is life threatening,
b) results in permanent impairment of a body function or permanent damage to a body structure, or
c) necessitates medical or surgical intervention to prevent permanent impairment of a body function or permanent damage to a body structure
[FDA 21CFR803]
serviceoutput of an organization with at least one activity necessarily performed between the organization and the customer
Note 1: The dominant elements of a service are generally intangible.
Note 2: Service often involves activities at the interface with the customer to establish customer requirements as well as upon delivery of the service and can involve a continuing relationship such as banks, accountancies or public organizations, e.g. schools or hospitals.
Note 3: Provision of a service can involve, for example, the following:
— an activity performed on a customer-supplied tangible product (e.g. a car to be repaired);
— an activity performed on a customer-supplied intangible product (e.g. the income statement needed to prepare a tax return);
— the delivery of an intangible product (e.g. the delivery of information in the context of knowledge transmission);
— the creation of ambience for the customer (e.g. in hotels and restaurants);
Note 4: A service is generally experienced by the customer.
[ISO 9000:2015]
servicing processprocess of combining various inputs in order to maintain or repair a medical device such that it is ready for use
severitymeasure of the possible consequences of a hazard
[ISO 14971:2019]
single-use devicea device that is intended for one use, or on a single patient during a single procedure
[OLRC 21USC9]
single-use devicea device that is intended to be used on one individual during a single procedure
[EU MDR 2017/745]
single-use devicea device that is intended to be used during a single procedure
[EU IVDR 2017/746]
single-use medical devicemedical device labelled or intended to be used on one individual during a single procedure
[ISO 11139:2018]
softwareThe set of electronic instructions used to control the actions or output of a medical device, to provide input to or output from a medical device, or to provide the actions of a medical device. This definition includes software that is embedded within or permanently a component of a medical device, software that is an accessory to another medical device, or software that is intended to be used for one or more medical purposes that performs these purposes without being part of a hardware medical device.
[FDA guidance: Deciding When to Submit...(2014)]
software as a medical device (SaMD)software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device
Notes:
• SaMD is a medical device and includes in-vitro diagnostic (IVD) medical device.
• SaMD is capable of running on general purpose (non-medical purpose) computing platforms
• “without being part of” means software not necessary for a hardware medical device to achieve its intended medical purpose;
• Software does not meet the definition of SaMD if its intended purpose is to drive a hardware medical device.
• SaMD may be used in combination (e.g., as a module) with other products including medical devices;
• SaMD may be interfaced with other medical devices, including hardware medical devices and other SaMD software, as well as general purpose software
• Mobile apps that meet the definition above are considered SaMD.
[IMDRF/SaMD WG/N10:2013, IMDRF/SaMD WG/N12:2014]
software itemany identifiable part of a computer program, i.e., source code, object code, control code, control data, or a collection of these items
Note: Three terms identify the software decomposition. The top level is the software system. The lowest level that is not further decomposed is the software unit. All levels of composition, including the top and bottom levels, can be called software items. A software system, then, is composed of one or more software items, and each software item is composed of one or more software units or decomposable software items. The responsibility is left to the manufacturer to provide the granularity of the software items and software units.
[IEC 62304:2015]
software systemintegrated collection of software items organized to accomplish a specific function or set of functions
[IEC 62304:2015]
software unitsoftware item that is not subdivided into other items
Note: The granularity of software units is defined by the manufacturer
[IEC 62304:2015]
specificationany requirement with which a product, process, service, or other activity must conform
[FDA 21CFR820]
specification developer[organization that] develops specifications for a device that is distributed under the establishment's own name but performs no manufacturing. This includes establishments that, in addition to developing specifications, also arrange for the manufacturing of devices labeled with another establishment’s name by a contract manufacturer.
[FDA website: Who Must Register...]
stakeholderperson or organization that can affect, be affected by, or perceive themselves to be affected by a decision or activity
Note: The term “interested party” can be used as an alternative to “stakeholder”.
[ISO 31000:2018]
state of the art (SOTA)developed stage of technical capability at a given time as regards products, processes and services, based on the relevant consolidated findings of science, technology and experience
Note: The state of the art embodies what is currently and generally accepted as good practice in technology and medicine. The state of the art does not necessarily imply the most technologically advanced solution. The state of the art described here is sometimes referred to as the “generally acknowledged state of the art”.
[ISO 14971:2019]
sterilefree from viable microorganisms
[ISO 11139:2018]
sterile medical devicemedical device intended to meet the requirements for sterility
Note: The requirements for sterility of a medical device can be subject to applicable regulatory requirements or standards.
[ISO 13485:2016]
sterilizationvalidated process used to render a product free from viable microorganisms
Note: In a sterilization process, the nature of microbial inactivation is exponential and thus the survival of a microorganism on an individual item can be expressed in terms of probability. While this probability can be reduced to a very low number, it can never be reduced to zero.
[ISO 11139:2018]
stock recoverythe correction or removal of a device that has not been marketed or that has not left the direct control of the manufacturer, i.e., the device is located on the premises owned, or under the control of, the manufacturer, and no portion of the lot, model, code, or other relevant unit involved in the corrective or removal action has been released for sale or use
[FDA 21CFR806]
subjecta human who participates in an investigation, either as an individual on whom or on whose specimen an investigational device is used or as a control. A subject may be in normal health or may have a medical condition or disease.
[FDA 21CFR812]
subjecthuman who participates in a clinical performance study or whose specimen is used in the study
Note: Depending on the study, a subject can be either a healthy individual or a patient.
[ISO 20916:2019]
subjectan individual who participates in a clinical investigation
[EU MDR 2017/745]
subjectan individual who participates in a performance study and whose specimen(s) undergo
in vitro examination by a device for performance study and/or by a device used for control purposes
[ISO 20916:2019]
subjectindividual who is or becomes a participant in a clinical investigation, either as a recipient of the investigational device or a comparator
Note: This includes healthy volunteers.
[ISO 14155:2020]
substantial equivalenceSubstantial equivalence means that the new device is at least as safe and effective as the predicate. A 510(k) requires demonstration of substantial equivalence to another legally U.S. marketed device.
Note 1: A device is substantially equivalent if, in comparison to a predicate it: has the same intended use as the predicate; and has the same technological characteristics as the predicate; or has the same intended use as the predicate; and has different technological characteristics and does not raise different questions of safety and effectiveness; and the information submitted to FDA demonstrates that the device is at least as safe and effective as the legally marketed device.
Note 2: A claim of substantial equivalence does not mean the new and predicate devices must be identical. Substantial equivalence is established with respect to intended use, design, energy used or delivered, materials, chemical composition, manufacturing process, performance, safety, effectiveness, labeling, biocompatibility, standards, and other characteristics, as applicable.
Note 3: A device may not be marketed in the U.S. until the submitter receives a letter declaring the device substantially equivalent. If FDA determines that a device is not substantially equivalent, the applicant may: resubmit another 510(k) with new data, request a Class I or II designation through the De Novo Classification process, file a reclassification petition, or submit a premarket approval application (PMA).
[FDA website: Premarket Notification 510(k)]
traceabilityability to trace the history, application or location of an object
[ISO 9000:2015]
trademark (TM)a word, name, symbol, or device that is used in trade with goods to indicate the source of the goods and to distinguish them from the goods of others
Note: A servicemark is the same as a trademark except that it identifies and distinguishes the source of a service rather than a product. The terms “trademark” and “mark” are commonly used to refer to both trademarks and servicemarks.
[USPTO]
unique device identifier (UDI)an identifier that adequately identifies a device through its distribution and use by meeting the requirements of 830.20 of this chapter
[FDA 21CFR801, FDA 21CFR803, FDA 21CFR806, FDA 21CFR810, FDA 21CFR814, FDA 21CFR820, FDA 21CFR821, FDA 21CFR822, FDA 21CFR830]
unique device identifier (UDI)a series of numeric or alphanumeric characters that is created through internationally accepted device identification and coding standards and that allows unambiguous identification of specific devices on the market
[EU MDR 2017/745, EU IVDR 2017/746]
US manufacturer of export only devices[organization that] manufactures medical devices that are not sold in the US and are manufactured solely for export to foreign countries
[FDA website: Who Must Register...]
usabilitycharacteristic of the user interface that facilitates use and thereby establishes effectiveness, efficiency and user satisfaction in the intended use environment
Note: All aspects of usability, including effectiveness, efficiency and user satisfaction, can either increase or decrease safety.
[IEC 62366-1:2015]
usability engineering (UE)application of knowledge about human behaviour, abilities, limitations, and other characteristics to the design of medical devices (including software), systems and tasks to achieve adequate usability
Note 1: Achieving adequate usability can result in acceptable risk related to use.
[IEC 62366-1:2015]
Note 2: Also known as human factors engineering (HFE)
usability engineering file (UEF)set of records and other documents that are produced by the usability engineering process
[IEC 62366-1:2015]
usability testmethod for exploring or evaluating a user interface with intended users within a specified intended use environment
[IEC 62366-1:2015]
use environmentactual conditions and setting in which users interact with the medical device
Note: The conditions of use or attributes of the use environment can include hygienic requirements, frequency of use, location, lighting, noise, temperature, mobility, and degree of internationalization.
[IEC 62366-1:2015]
use erroruser action or lack of user action while using the medical device that leads to a different result than that intended by the manufacturer or expected by the user
Note 1: Use error includes the inability of the user to complete a task.
Note 2: Use errors can result from a mismatch between the characteristics of the user, user interface, task, or use environment.
Note 3: USERS might be aware or unaware that a use error has occurred.
Note 4: An unexpected physiological response of the patient is not by itself considered use error.
Note 5: A malfunction of a medical device that causes an unexpected result is not considered a use error.
Note 6: The figure below shows the relationships of the types of use.
[IEC 62366-1:2015, ISO 14971:2019]
userany healthcare professional or lay person who uses a device
[EU MDR 2017/745, EU IVDR 2017/746]
usermost IVD medical devices have two users that need to be considered:
- a user who performs all or part of an examination (“analytical use”); and
- a clinician who receives, interprets and acts on the examination results (“clinical use”).
Note: In the case of IVD medical devices intended for self-testing, the patient can be the only user.
[ISO/TR 24971:2020]
user (operator)person interacting with (i.e. operating or handling) the medical device
Note 1: There can be more than one user of a medical device.
Note 2: Common users include clinicians, patients, cleaners, maintenance and service personnel.
[IEC 62366-1:2015]
user interfacemeans by which the user and the medical device interact
Note 1: Accompanying documentation is considered part of the medical device and its user interface.
Note 2: User interface includes all the elements of the medical device with which the user interacts including the physical aspects of the medical device as well as visual, auditory, tactile displays and is not limited to a software interface.
Note 3: For the purposes of this standard, a system of medical devices can be treated as a single user interface.
[IEC 62366-1:2015]
user needs (customer needs)functions and characteristics of the medical device necessary to address the needs of the medical device user, patient, and other customers
Note: "Other customers" include other people who interact with the medical device during use and stakeholders who do not directly interact with the medical device such as post-production monitoring personnel, administrative personnel, regulatory bodies, and business stakeholders.
user profilesummary of the mental, physical and demographic traits of an intended user group, as well as any special characteristics, such as occupational skills, job requirements and working conditions, which can have a bearing on design decisions
[IEC 62366-1:2015]
validationconfirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled
[FDA 21CFR820]
validationconfirmation, through the provision of objective evidence, that the requirements for a specific intended use or application have been fulfilled
[ISO 9000:2015]
verificationconfirmation, through the provision of objective evidence, that specified requirements have been fulfilled
Note 1: The objective evidence needed for a verification can be the result of an inspection or of other forms of determination such as performing alternative calculations or reviewing documents.
Note 2: The activities carried out for verification are sometimes called a qualification process.
Note 3: The word “verified” is used to designate the corresponding status.
[ISO 9000:2015, ISO 14971:2019]
verificationconfirmation by examination and provision of objective evidence that specified requirements have been fulfilled
[FDA 21CFR820, ISO 20916:2019]
voice of the customer (VOC)customer (market) research to determine the medical device intended use and user needs
